In silico identification and target prediction of micrornas in Sesame (sesamum indicum L.) expressed sequence tags
Received: April 17, 2018
Accepted: April 23, 2018
Published: April 28, 2018
Genet.Mol.Res. 17(2): gmr16039911
DOI: 10.4238/gmr16039911
Abstract
Sesame (Sesamum indicum L.), a member of the Pedaliaceae family, is one of the oldest oilseed crops. For its high oil content, it is known as the “queen of oilseeds”. MicroRNAs (miRNAs) represent a class of endogenous non-coding small RNAs that play important roles in multiple biological processes by degrading targeted mRNAs or repressing mRNA translation. Thousands of miRNAs have been identified in many plant species by computational methods, whereas there is no report of miRNAs in S. indicum till date. In present study, previously known plant miRNAs were BLASTed against the Expressed Sequence Tag (EST) database of Sesame genes. The aligned miRNA hits were further aligned to protein database and BLASTX was carried out to remove protein coding primary miRNAs. The non-coding precursor miRNAs were subjected to online MFold server in order to predict their secondary structures. After applying the filtering criteria, a total of 12 potential miRNAs belonging to 6 miRNAs families were detected. 203 unique miRNAs: target pairs were predicted online by psRNATarget web server. Most of the targets were found to encode transcription factors or enzymes that participate in the regulation of development, growth, metabolism, and other physiological processes and stress response.
Introduction
Sesame (Sesamum indicum L.), a member of the Pedaliaceae, is a diploid (2n = 26) dicotyledon and one of the oldest oil seed crops. The Sesame seeds have been considered as the “queen of oilseeds” for its high oil content (55–58%). It is cultivated mainly in the tropical and subtropical regions of Asia, Africa and Southern America. India is the major producer of Sesame seeds followed by Sudan, China, Myanmar and Tanzania. These top five countries account for 80% of world production. Sesame seeds are important source of oil (44-58%), protein (18-25%), and carbohydrates (13.5%). (Cheung et al., 2007) (Anila kumar et al., 2010).
MicroRNAs (miRNAs) are endogenous ~22 nucleotides-long RNAs that play important regulatory roles in animals and plants by targeting messenger RNAs (mRNAs), for cleavage or translational repression. miRNAs comprise one of the most abundant classes of gene regulatory molecules in multicellular organisms. (Jonas and Izaurralde, 2015). In plants, miRNAs control crucial biological processes like flowering, polarity, nutrient homeostasis, phase-change, biotic and abiotic stress responses by influencing the output of many protein-coding genes (Dugas and Bartel, 2004). All miRNA precursors have a well-predicted stem loop hairpin structure, and this fold-back hairpin structure has a low free energy. Many miRNAs are evolutionarily conserved. Different computational miRNA finding strategies have been developed based on these characteristics of miRNAs (Zhang et al., 2005).
There are two major approaches for identification of miRNAs in plants: experimental approaches and computational approaches. Computational approaches are faster and more affordable than the experimental approaches. Expressed Sequence Tags (ESTs) provide a powerful tool for identification of miRNAs that are conserved among various plant species. These methods are used to study the conservation and evolution of miRNAs among different species. The miRNA identification and target prediction by computational methods and recent developments have increased the speed of generating new strategies making the future of miRNA target prediction promising. (Zhang et al., 2006).
The present study was carried out to investigate the information about Sesame miRNAs by predicting miRNAs from Sesame est database and predicting their secondary structures using computational approach. the potential targets for predicted mirnas were also predicted using computational methods.
Material and Methods
Sequence and computational requirements
A total of 44905 Expressed Sequence Tag (EST) of S. indicum, deposited at Genbank database of the National Center for Biotechnology Information (NCBI) was downloaded. Protein coding sequences of S. indicum were downloaded from Protein database of NCBI (http://www.ncbi.nlm.nih.gov/).
A total of 8220 plant miRNA sequences were retrieved from the previously deposited miRNA from the publically available miRNA database miRBase, version 21. (http://www.mirbase.org/) (Griffith-Jones et al., 2008).
Prediction of miRNAs
Redundancy among the miRNAs and ESTs was excluded manually. The known plant miRNA sequences were subjected to the BLAST search for Sesame homologs of miRNAs against EST database. Local BLAST search was performed with BioEdit version 7.0.5. (Hall, 1999). The expectation value was kept at 0.001 and rest of the parameters were kept at default. The results were transferred to separate data sheets. The miRNAs having more than 3nt mismatches, less than 90% identity and less than 18 alignment length were removed.
Protein BLAST
As plant miRNAs are unlikely to be located in protein coding genes, the protein blast search was performed by BLASTX of BioEdit (Hall, 1999). All the protein coding sequences from aligned miRNAs were removed and the remaining sequences were most likely to be potential miRNAs.
Prediction of secondary structure
After inspection of all hit sequences, a new database was formed from the aligned miRNA sequences, sequence with 100 nucleotides upstream and downstream of the first and last residues of the mature miRNA were extracted. These sequences were treated as precursor microRNAs (pre-miRNAs). The secondary structures were predicted online using RNA folding form of The Mfold Web Server (Zuker, 2003). (https://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form).
The secondary structures were checked to meet the criteria by Zhang et al., (2005). If it met all of them, the sequence was selected and the 5’ and 3’ ends were determined.
i. Pre-miRNA sequence can fold into an appropriate stem-loop hairpin secondary structure.
ii. It contained ~22 nt mature miRNA sequence within one arm of the hairpin.
iii. Minimal folding free energy MFEâ�?¦-20 kcal/mol.
iv. 30–70% A+U content
v. Predicted mature miRNAs had no more than six mismatches with the opposite miRNA* sequence in the other arm
vi. Maximum size of 3 nucleotides for a bulge in the miRNA sequence.
vii. No loop or break in miRNA sequences was allowed.
All the potential miRNA sequences were renamed for Sesamum indicum according to miRBase nomenclature guideline (Griffiths-Jones et al., 2006).
miRNA target prediction
Based on the newly identified potential miRNA sequences, their potential targets were predicted using online server psRNATarget against nucleotide sequences of S. indicum as target transcript sequences. (http://plantgrn.noble.org/psRNATarget/) (Dai and Zhao, 2011).
Results
Prediction of miRNA
A comparative genomic approach was carried out for initial prediction of miRNAs. The hits having <90% similarity and <18 nucleotides length were removed and 132 hits were remained. These miRNA sequences were further subjected to protein BLASTX. 5 EST sequences were found to be protein coding. So, these were eliminated and the remaining 127 pre-miRNA sequences were analyzed for prediction of secondary structures.
Secondary structure prediction
127 pre-miRNA sequences were carried on for secondary structure prediction by M-Fold software. The miRNAs were assessed for their proper folding structure, Minimum Folding Free Energy (MFE), number of nucleotides etc. The predicted structures are shown in Figure 1. The other outputs related to structures are depicted in Table 1 and Table 2.

Figure 1: Pre-miRNA sequences carried on for secondary structure prediction by M-Fold software
| Family | miRNA | Sequence | Length | Location | %A+U | MFE (Kcal/mol) | MFEi |
|---|---|---|---|---|---|---|---|
| 156/157 | sin-miR156f | uugacagaagagagagagcaca | 22 | 5' | 54.55 | 66.5 | 1.46 |
| sin-miR156h | ugacagaagagagagagca | 19 | 5' | 52.63 | 66.5 | 1.4 | |
| sin-miR156j | ugacagaagagagagagcac | 20 | 5' | 50 | 66.5 | 1.33 | |
| sin-miR156k | ugacagaagagagagagcaca | 21 | 5' | 52.38 | 68.23 | 1.43 | |
| sin-miR157a-5p | uugacagaagagagagagcac | 21 | 5' | 57.14 | 77.1 | 1.8 | |
| 396 | sin-miR396a-5p | uuccacagcuuucuugaacug | 21 | 5' | 57.14 | 72.8 | 1.7 |
| 529 | sin-miR529 | agaagagagagagcacagc | 19 | 5' | 42.11 | 66.5 | 1.15 |
| sin-miR529a | gaagagagagagcacagccc | 21 | 5' | 42.86 | 74.68 | 1.31 | |
| sin-miR529d | agaagagagagagcacagccc | 19 | 5' | 47.37 | 66.5 | 1.26 | |
| 5658 | sin-miR5658 | augaugaugaugaugauga | 19 | 5' | 68.42 | 30 | 0.95 |
| 8030 | sin-miR8030-3p | aaaccaaaucaacccaaau | 19 | 3' | 68.42 | 39.73 | 1.26 |
| 8138 | sin-miR8138 | uaaagaugguaacaaagg | 18 | 3' | 66.67 | 43.15 | 1.29 |
Table 1: The characteristics of predicted miRNAs including family, sequences, length, location of miRNAs in precursor
| miRNA | Targets | Inhibition | Target Description |
|---|---|---|---|
| sin-miR156f | XM_011073548.2 | Cleavage | PREDICTED: Sesamumindicum 1-deoxy-D-xylulose 5-phosphate reductoisomerase, chloroplastic (LOC105157212), mRNA |
| sin-miR156f | XM_011082778.2 | Cleavage | PREDICTED: Sesamumindicum protein TPX2 (LOC105164177), transcript variant X1, mRNA |
| sin-miR156f | XM_011082779.2 | Cleavage | PREDICTED: Sesamumindicum protein TPX2 (LOC105164177), transcript variant X2, mRNA |
| sin-miR156h | XM_011073548.2 | Translation | PREDICTED: Sesamumindicum 1-deoxy-D-xylulose 5-phosphate reductoisomerase, chloroplastic (LOC105157212), mRNA |
| sin-miR156h | XM_011101719.2 | Cleavage | PREDICTED: Sesamumindicum classical arabinogalactan protein 4-like (LOC105178272), mRNA |
| sin-miR156j | XM_011098026.2 | Translation | PREDICTED: Sesamumindicumsquamosa promoter-binding protein 1 (LOC105175555), mRNA |
| sin-miR157a-5p | XM_020695985.1 | Cleavage | PREDICTED: Sesamumindicumsquamosa promoter-binding-like protein 12 (LOC105168427), transcript variant X1, mRNA |
| sin-miR396a-5p | XM_011079770.2 | Cleavage | PREDICTED: Sesamumindicum growth-regulating factor 1 (LOC105161914), mRNA |
| sin-miR529a | XM_011093891.2 | Cleavage | PREDICTED: Sesamumindicum ATP-citrate synthase alpha chain protein 2 (LOC105172457), mRNA |
| sin-miR529a | XM_020695985.1 | Cleavage | PREDICTED: Sesamumindicumsquamosa promoter-binding-like protein 12 (LOC105168427), transcript variant X1, mRNA |
| sin-miR5658 | XM_020692989.1 | Cleavage | PREDICTED: Sesamumindicum ABC transporter B family member 19-like (LOC105158093), mRNA |
| sin-miR5658 | XM_011073794.2 | Cleavage | PREDICTED: Sesamumindicum alpha-galactosidase (LOC105157383), mRNA |
| sin-miR5658 | XM_011077608.2 | Cleavage | PREDICTED: Sesamumindicumauxin-responsive protein SAUR32 (LOC105160291), mRNA |
| sin-miR5658 | XM_011099043.2 | Cleavage | PREDICTED: Sesamumindicum BAG family molecular chaperone regulator 1-like (LOC105176294), mRNA |
| sin-miR5658 | XM_011076179.2 | Cleavage | PREDICTED: SesamumindicumBEL1-like homeodomain protein 4 (LOC105159201), mRNA |
| sin-miR5658 | XM_011092565.2 | Cleavage | PREDICTED: Sesamumindicumbrefeldin A-inhibited guanine nucleotide-exchange protein 2 (LOC105171444), mRNA |
| sin-miR5658 | XM_011085319.2 | Cleavage | PREDICTED: SesamumindicumbZIP transcription factor 44 (LOC105166092), mRNA |
| sin-miR5658 | XM_011080551.2 | Cleavage | PREDICTED: SesamumindicumbZIP transcription factor TGA10 (LOC105162512), mRNA |
| sin-miR5658 | XM_011081773.2 | Cleavage | PREDICTED: Sesamumindicum casein kinase 1-like protein 10 (LOC105163434), mRNA |
| sin-miR5658 | XM_011072757.2 | Cleavage | PREDICTED: Sesamumindicumcyclin-D1-1 (LOC105156585), mRNA |
| sin-miR8030-3p | XM_011074620.2 | Translation | PREDICTED: Sesamumindicum lysine-specific demethylaseREF6 (LOC105158021), transcript variant X1, mRNA |
| sin-miR8030-3p | XM_011096846.2 | Cleavage | PREDICTED: Sesamumindicum probable WRKY transcription factor 70 (LOC105174673), mRNA |
| sin-miR8030-3p | XM_011094724.2 | Cleavage | PREDICTED: Sesamumindicum transcription factor bHLH13-like (LOC105173075), transcript variant X1, mRNA |
Table 2: miRNA and their predicted targets with their description
All MFEs were expressed as negative kcal/mol. Adjusted MFE (AMFE) represented the MFE of 100 nucleotides. It was calculated by following equation.
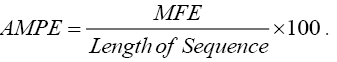
The minimal folding free energy index (MFEI) was calculated by the equation:
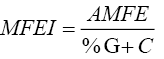
The MFE ranged from -30 to -77.1 Kcal/mol. The average value was found to be -61.52 Kcal/mol. Most of the miRNAs had MFEI greater than 1. The predicted miRNAs varied from 18-22 nucleotides length with an average of 20 nucleotides. miRNAs bind more strongly to certain proteins as they have a higher(A+U) content compared to other RNAs. The %A+U contents were lying between 42.11 to 68.42% with an average value of 54.97%. miRNAs were located at both the 5’ and 3’ arm of precursor stem-loop structures. Mature miRNA sequences have been reported to be evenly located on both the arms of the stem-loop hairpin structures of potential pre-miRNAs (Gorodkin et al., 2006). In the present study, more mature miRNAs were located in the 5’ end of the pre-miRNA than were located in the 3’ end.
After applying all the filtering criteria, a total of 12 miRNAs belonging to 6 different families were predicted.
miRNA target prediction
Targets of these miRNAs were predicted by online server named psRNATarget. Gene sequences of Sesamum indicum were used as database to predict the targets. Total of 203 unique targets were predicted for all the miRNAs. Some of the miRNAs and their targets are shown in table 2. The mode of inhibition of predicted miRNAs were either cleaving the target or translation inhibition. In most of the cases, the inhibition was found to be the cleavage of targets. Only 5 out of 203 predicted targets were inhibited by translation. The highest targets were predicted for sin-miR5658 family with 64 targets and the lowest targets were predicted for sin-miR8030. The number of targets predicted for each family was irrespective of the number of members in it.
Discussion
The nature of sequences conservation across different species for most of the known miRNAs is well known. Most miRNAs have shown to be conserved among related species and homologs were even found among distantly related species. Due to the high degree of sequence identity within families of mature miRNAs of plants, the number of hits was reduced to ~90%. When compared to the number of miRNA reported in other plant species and on the basis of the principle of prediction of one mature miRNA for every 10,000 EST sequence, at least 5 mature should have been identified from Sesame . The small number miRNA families identified may be due to fewer EST sequences available for S. indicum (44,905) than that of other plant species (Axtell and Bartel., 2005).
Formation of the stem-loop hairpin secondary structure is a critical step in miRNA maturation and one of the most important characteristics of pre-miRNAs. However, a stem-loop hairpin structure is not a unique characteristic of miRNA. Other RNAs (mRNA, rRNA, and tRNA) can also form similar hairpin structures. Thus, a potential stem-loop hairpin structure containing the ~22-nt mature miRNA sequence within one arm of the hairpin is a basic precondition for predicting and annotating new miRNAs or miRNA homologs. The MFEI can be easily used to distinguish miRNA from other non-coding and coding RNAs. The MFEI is a unique criterion to designate miRNAs. When the MFEI is more than 0.85, the sequence is most likely to be miRNA.
The prediction of targets for the identified miRNAs was expected to help us for clarification of the important function and regulation of these novel miRNAs in S. indicum. Most plant miRNAs are perfectly or near-perfectly complementary with their targets (Schwab et al., 2005). For this reason, searching potential miRNA targets by blasting the mature miRNA sequences against the nucleotide or genome database is considered a reliable technique. Nevertheless, several studies have demonstrated that many target genes have 1 to 4 nucleotide mismatches with mature miRNA sequences (Xie et al., 2010).
The predicted potential targets belonged to several gene families, and had multiple biological functions. Most of the targets were transcription factors. In addition to the transcription factors, the predicted targeted genes were involved in a broad range of biological processes, such as abiotic stress response, metabolism, transportation, disease resistance, and signal transduction. It was reported by Zhang et al., (2005), that 26% of EST contigs containing miRNAs are related to different biotic or abiotic environmental stresses, suggesting that environmental stress may play an important role in miRNA gene expression in plants.
Highly conserved S. indicum miRNAs also had conserved miRNA target sites on specific target genes, which was also observed in previous studies of other plants (Frazier et al., 2010). For example, it has been established that the plant-specific transcription factor, squamosa promoter binding protein, is involved in regulating changes during the early flower development and vegetative phase. In addition, it has been widely accepted that this transcription factor is a conserved target of the miR156 family in plants (Yin et al., 2008). In present study, 16 different squamosa promoter binding protein-encoding genes were identified as the targets of miR156/157 in S. indicum. Other than these cationic amino acid transporter, TPX2 micro-tubule binding protein and F-box protein involved in protein ubiquitination were also predicted for miR156/157. Different types of auxin response factors (ARFs) that are known to be engaged in signal transduction were predicted. This result can be compared to other studies in Arabidopsis. NAC domain containing proteins are known to be involved in plant morphogenesis, auxin responses and root development were predicted. (Zhang et al., 2006 and Mallory et al., 2005).
miR5658 has highest predicted targets. The targets included various transcription factors, proteins involved in signal transduction, molecular chaperone regulator, growth regulators, protein ligases, WRKY transcription factor, argonaute protein, etc. The earlier reports support this result showing more numbers of targets predicted by miR5658 (Han et al., 2013; Li et al., 2014). miR5658 targeted zinc finger transcription factors are a superfamily of proteins involved in numerous activities of plant growth and development, and are also known to regulate resistance mechanisms for various biotic and abiotic stresses (Giri et al., 2011). The basic/helixloop-helix (bHLH) transcription factors that control cell proliferation and cell lineage establishment are potential targets for miR5658. miR5658 also target the mRNA of homeobox-leucine zipper transcription factor, which is reported to regulate leaf polarity and vascular differentiation via the miR165/166 family (Jung and Park, 2007).
Many studies have shown that plant miRNAs are involved in biotic and abiotic stress (Xie et al., 2010; Gao et al., 2011; Sunkar et al., 2012). In present studies it was also identified the stress-related proteins targeted by S. indicum miRNAs. In sunflower, when plant exposed to high temperatures, a WRKY TF exhibited inverse correlation with miR5658. High level of miR5658 was observed in older leaves in contrast to the distal portion where the expression was low (Giacomelli et al., 2012). In rice treated with arsenic, miR5658 was downregulated which resulted in the upregulation of its target, WRKY TF (Liu and Zhang, 2012).
miR5658 targeted the heat shock proteins that are expressed in response to heat stress, which indicates the important role of these miRNAs during heat stress. Most plant resistance genes (R genes) encode nucleotide binding site leucine-rich repeat (NBS-LRR) proteins (McHale et al., 2006). The NBS-LRR resistance protein was predicted to be the target of the S. indicum miR5658 family.
Summary and Conclusion
Numbers of microRNAs have been predicted through computational methods for various plant but for Sesame , no miRNAs have been reported till date. Availability of good set of EST database provided an opportunity to peep into gene regulation and prediction of miRNAs. Total of 12 microRNA belonging to 6 different families were predicted using computational methods. These computational methods are cost-effective and faster than other conventional methods. There is a well-established fact that a mature miRNA is derived from its precursor miRNA and its structure can be used as a search template. A pre-RNA forms a short stable extended stem-loop structure or a hairpin structure, with continuous helical pairing and a few internal bulges. There are strict filtering criteria to distinguish miRNAs from other small RNAs. So, the prediction of secondary structure proved to be the most important filtering criteria. More than 70% pre-miRNAs were not fulfilling all the filtering criteria. For 12 miRNAs, 203 non-duplicated unique targets were predicted. Most of these target genes were predicted to be involved in plant development, signal transduction, metabolic pathways, disease resistance, and environmental stress response. Thus the identification of the novel miRNAs in Sesamum indicum is foreseen to provide baseline information for further research about the biological functions and evolution of miRNAs in S. indicum
About the Authors
Corresponding Author
Halak Joshi
Department of Biotechnology, Junagadh Agricultural University, Junagadh-362001, India
- Email:
- halakjoshi@gmail.com
References
- Anilakumar KR, Pal A, Khanum F, and Bawa (2010). Nutritional, medicinal and industrial uses of Sesame(SesamumindicumL.)seeds - an overview.Agriculturae Conspectus Scientificus.,75(4):159-168.
- Axtell MJ and Bartel DP (2005) Antiquity of MicroRNAs and their targets in land plants.The Pl. Cell, 17 (6):1658–1673.https://doi.org/10.1105/tpc.105.032185
- Cheung SC, Szeto YT and Benzie IF (2007) Antioxidant protection of edible oils.Pl. Foods Hum.Nutr.,62 (1):39-42.https://doi.org/10.1007/s11130-006-0040-6
- DaiX and ZhaoPX(2011)psRNATarget: a plant small RNA target analysis server Nucleic A. Res., 39: 155-159.https://doi.org/10.1093/nar/gkr319
- Dugas DV and Bartel B (2004) MicroRNA regulation of gene expression in plants.Cur.Opin. Plant Biol.7: 512–520.
- Frazier TP, Xie F, Freistaedter A, Burklew CE, et al.,(2010).Identification and characterization of microRNAs and their target genes in tobacco (Nicotianatabacum).Planta, 232 (6):1289–1308.https://doi.org/10.1007/s00425-010-1255-1
- Gao P,Bai X, Yang L and Lv D (2011)osa-MIR393: a salinity- and alkaline stress-related microRNA gene. Mol. Biol. Rep. 38 (1): 237-242.https://doi.org/10.1007/s11033-010-0100-8
- GiacomelliJI, Weigel D, Chan RL andManavella PA (2012).Role of recently evolved miRNA regulation of sunflower HaWRKY6 in response to temperature damage.New Phytol.,195 (4):766–773.https://doi.org/10.1111/j.1469-8137.2012.04259.x
- Giri J, Vij S, DansanaPK,Tyagi AK (2011) Rice A20/AN1 zinc-finger containing stress-associated proteins (SAP1/11) and a receptor-like cytoplasmic kinase (OsRLCK253) interact via A20 zinc-finger and confer abiotic stress tolerance in transgenic Arabidopsis plants. New Phytol.191 (3): 721-732.https://doi.org/10.1111/j.1469-8137.2011.03740.x
- Gorodkin J, HavgaardJH, Enstero, MSawera, et al., (2006). Comput.Biol. Chem.30: 249–254.
- Griffiths-JonesS, Grocock RJ, DongenSV, Bateman A, et al.,(2006). miRBase: microRNA sequences, targets and gene nomenclature. Nucleic A. Res.,34: 140-144.https://doi.org/10.1093/nar/gkj112
- Griffiths-Jones S, Saini HK, DongenSV and Enright AJ(2008) miRBase: tools for microRNA genomics. Nucleic A. Res., 36: 154-158.https://doi.org/10.1093/nar/gkm952
- Hall TA (1999)BioEdit: a user friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic. A. Symp.,41:95-98.
- Han J, Xie H, Kong ML, Sun QP, et al., (2013). Computational identification of miRNAs and their targets in Phaseolus vulgaris.Genet. Mol. Res.13 (1): 310-322.https://doi.org/10.4238/2014.january.17.16
- Jonas S and Izaurralde E (2015).Towards a molecular understanding of microRNA-mediated gene silencing.Nature Reviews Genetics,16 (7):421-433.https://doi.org/10.1038/nrg3965
- Li X, Hou Y, Zhang L, Zhang W, et al., (2014). Computational identification of conserved microRNAs and their targets from expression sequence tags by blueberry. Pl. Signal.Behav.9 (9):29462.https://doi.org/10.4161/psb.29462
- Liu Q and Zhang H (2012).Molecular identification and analysis of arsenite stress-responsive miRNAs in rice.J. Agric. Food Chem.60 (26):6524–6536.https://doi.org/10.1021/jf300724t
- MalloryAC,Bartel DP,Bartel B (2005). MicroRNA-Directed Regulation of Arabidopsis AUXIN RESPONSE FACTOR17 Is Essential for Proper Development and Modulates Expression of Early Auxin Response Genes. The Plant Cell, 17 (5):1360–1375.https://doi.org/10.1105/tpc.105.031716
- McHaleL, Tan X, Koehl P, and MichelmoreRW(2006). Plant NBS-LRR proteins: adaptable guards. Genome Biol.7: 212.
- Schwab R, PalatnikJF, Riester M, Schommer C, et al., (2005). Specific Effects of MicroRNAs on the Plant Transcriptome.Develop. Cell, 8 (4): 517–527.https://doi.org/10.1016/j.devcel.2005.01.018
- Sunkar R., Li YF and Jagadeeswaran G (2012). Functions of microRNAs in plant stress responses. Trends Pl. Sci., 17:196-203.https://doi.org/10.1016/j.tplants.2012.01.010
- Xie F, Frazier TP and Zhang BH(2010). Identification and characterization of microRNAs and their targets in the bioenergy plant switchgrass (Panicumvirgatum). 232 (2): 417-434.https://doi.org/10.1007/s00425-010-1182-1
- Zhang BH, Pan XP, Wang QL, Cobb GP et al., (2005). Identification and characterization of new plant microRNAs using EST analysis.Cell Res.,15(5):336-360.https://doi.org/10.1038/sj.cr.7290302
- Zhang B, Pan X, Cobb GP and Anderson TA (2006) Plant microRNA: A small regulatory molecule with big impact. Develop. Biol., 289 (1):3 – 16.https://doi.org/10.1016/j.ydbio.2005.10.036
- ZukerM (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic A. Res, 31(3):3406-3415.https://doi.org/10.1093/nar/gkg595
Keywords:
Download:
Full PDF- Share This