Abstract
Protein-protein interaction network and significant gene analysis of osteoporosis
Author(s): X.M. Wu, X. Ma, C. Tang, K.N. Xie, J. Liu, W. Guo, Y.L. Yan, G.H. Shen and E.P. LuoThis study used DNA microarray data to identify differentially expressed genes of osteoporosis and provide useful information for treatments of the disease. We downloaded gene expression data of Osteoporosis GSE35956 from the Gene Expression Omnibus database, which included five normal and five osteoporosis samples. We then identified the differentially expressed genes between normal and disease samples using the R language software, and constructed the protein interaction network. DAVID was used to perform the biological process enrichment and KEGG pathway cluster analyses. We used the Cytoscape plug-in unit, Cluster ONE, to perform cluster module analysis to find hub proteins of the network module and to analyze their Gene Ontology (GO) functions. A total of 294 genes were found to be differentially expressed between normal and disease samples, which were used to construct the differential gene-protein interaction network. GO function analysis revealed that the genes’ functions were mainlyinvolved in the intracellular signaling cascade. KEGG pathway analysis suggested that the main metabolic pathways of these genes were those of cancer: the neurotrophin/T cell/Fc epsilon RI/B cell/ ErbB/p53 signaling pathway, the cell cycle pathway, and the chronic myeloid leukemia pathway. Screening analysis of hub proteins revealed that KRT18 had the highest hub degree. In conclusion, we found differentially expressed genes related to osteoporosis. GO biological process enrichment and KEGG pathway enrichment analyses identified significant osteoporosis genes and their molecular functions. Finally, module analysis of hub proteins in interaction networks showed that cell death was one of the main biological processes of osteoporosis genes.
Impact Factor an Index

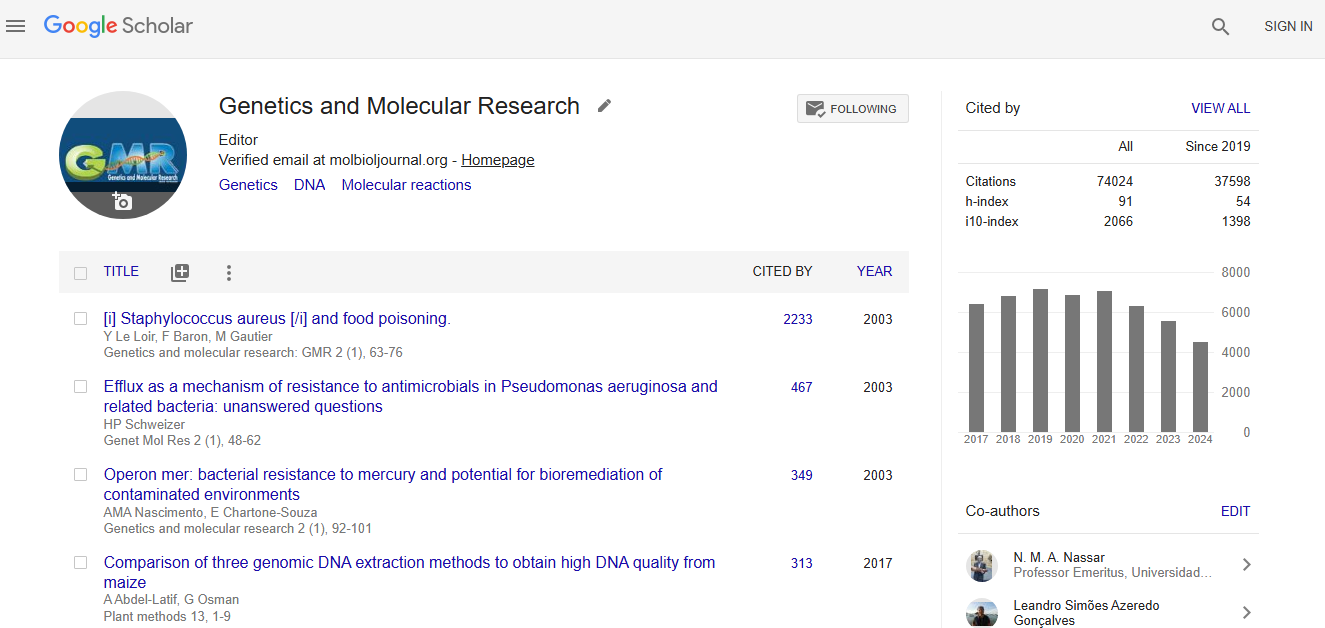
Google scholar citation report
Citations : 74024
Genetics and Molecular Research received 74024 citations as per google scholar report