Abstract
Genome-wide analysis of salinity-stress induced DNA methylation alterations in cotton (Gossypium hirsutum L.) using the Me-DIP sequencing technology
Author(s): X.K. Lu, N. Shu, J.J. Wang, X.G. Chen, D.L. Wang, S. Wang, W.L. Fan, X.N. Guo, L.X. Guo and W.W. YeCytosine DNA methylation is a significant form of DNA modification closely associated with gene expression in eukaryotes, fungi, animals, and plants. Although the reference genomes of cotton (Gossypium hirsutum L.) have been publically available, the salinity-stress-induced DNA methylome alterations in cotton are not well understood. Here, we constructed a map of genome-wide DNA methylation characteristics of cotton leaves under salt stress using the methylated DNA immunoprecipitation sequencing method. The results showed that the methylation reads on chromosome 9 were most comparable with those on the other chromosomes, but the greatest changes occurred on chromosome 8 under salt stress. The DNA methylation pattern analysis indicated that a relatively higher methylation density was found in the upstream2k and downstream2k elements of the CDS region and CG-islands. Almost 94% of the reads belonged to LTR-gspsy and LTR-copia, and the number of methylation reads in LTR-gypsy was four times greater than that in LTR-copia in both control and stressed samples. The analysis of differentially methylated regions (DMRs) showed that the gene elements upstream2k, intron, and downstream2k were hypomethylated, but the CDS regions were hypermethylated. The GO (Gene Ontology) analysis suggested that the methylated genes were most enriched in cellular processes, metabolic processes, cell parts and catalytic activities, which might be closely correlated with response to NaCl stress. In this study, we completed a genomic DNA methylation profile and conducted a DMR analysis under salt stress, which provided valuable information for the better understanding of epigenetics in response to salt stress in cotton.
Impact Factor an Index

Google scholar citation report
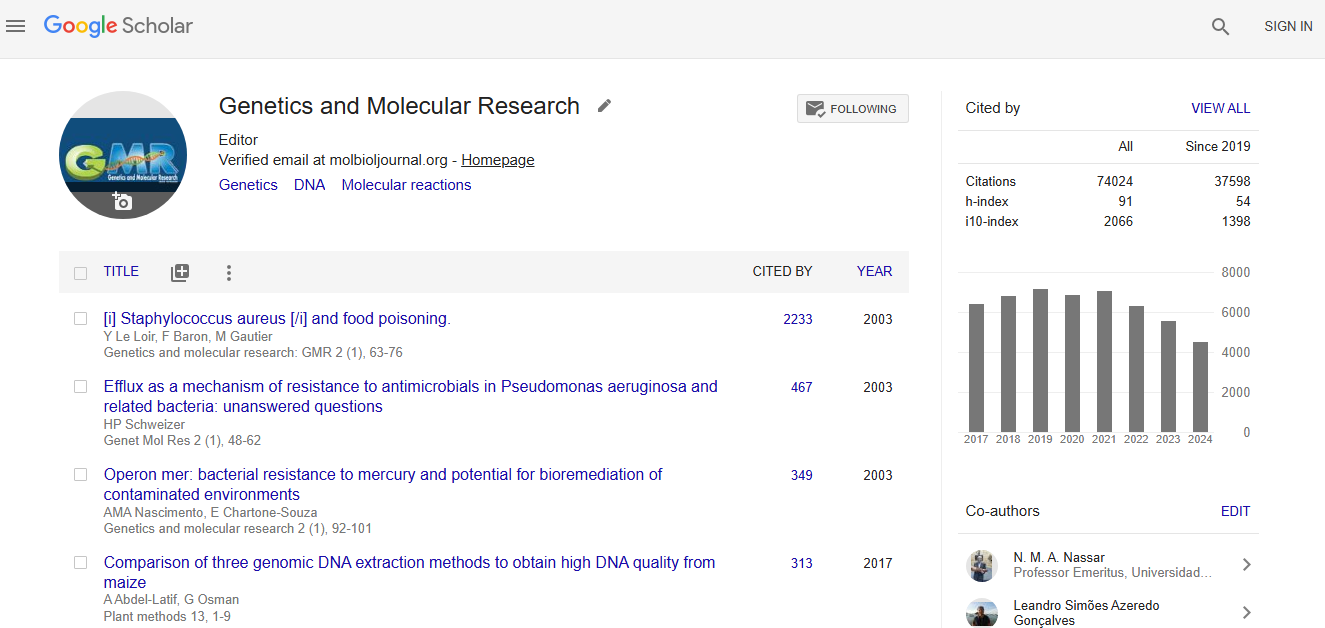
Citations : 74024
Genetics and Molecular Research received 74024 citations as per google scholar report