Abstract
E2F, HSF2, and miR-26 in thyroid carcinoma: bioinformatic analysis of RNA-sequencing data
Author(s): J.C. Lu and Y.P. ZhangIn this study, we examined the molecular mechanism of thyroid carcinoma (THCA) using bioinformatics. RNA-sequencing data of THCA (N = 498) and normal thyroid tissue (N = 59) were downloaded from The Cancer Genome Atlas. Next, gene expression levels were calculated using the TCC package and differentially expressed genes (DEGs) were identified using the edgeR package. A co-expression network was constructed using the EBcoexpress package and visualized by Cytoscape, and functional and pathway enrichment of DEGs in the co-expression network was analyzed with DAVID and KOBAS 2.0. Moreover, modules in the co-expression network were identified and annotated using MCODE and BiNGO plugins. Small-molecule drugs were analyzed using the cMAP database, and miRNAs and transcription factors regulating DEGs were identified by WebGestalt. A total of 254 up-regulated and 59 down-regulated DEGs were identified between THCA samples and controls. DEGs enriched in biological process terms were related to cell adhesion, death, and growth and negatively correlated with various small-molecule drugs. The co-expression network of the DEGs consisted of hub genes (ITGA3, TIMP1, KRT19, and SERPINA1) and one module (JUN, FOSB, and EGR1). Furthermore, 5 miRNAs and 5 transcription factors were identified, including E2F, HSF2, and miR-26. miR-26 may participate in THCA by targeting CITED1 and PLA2R1; E2F may participate in THCA by regulating ITGA3, TIMP1, KRT19, EGR1, and JUN; HSF2 may be involved in THCA development by regulating SERPINA1 and FOSB; and small-molecule drugs may have anti-THCA effects. Our results provide novel directions for mechanistic studies and drug design of THCA. In this study, we examined the molecular mechanism of thyroid carcinoma (THCA) using bioinformatics. RNA-sequencing data of THCA (N = 498) and normal thyroid tissue (N = 59) were downloaded from The Cancer Genome Atlas. Next, gene expression levels were calculated using the TCC package and differentially expressed genes (DEGs) were identified using the edgeR package. A co-expression network was constructed using the EBcoexpress package and visualized by Cytoscape, and functional and pathway enrichment of DEGs in the co-expression network was analyzed with DAVID and KOBAS 2.0. Moreover, modules in the co-expression network were identified and annotated using MCODE and BiNGO plugins. Small-molecule drugs were analyzed using the cMAP database, and miRNAs and transcription factors regulating DEGs were identified by WebGestalt. A total of 254 up-regulated and 59 down-regulated DEGs were identified between THCA samples and controls. DEGs enriched in biological process terms were related to cell adhesion, death, and growth and negatively correlated with various small-molecule drugs. The co-expression network of the DEGs consisted of hub genes (ITGA3, TIMP1, KRT19, and SERPINA1) and one module (JUN, FOSB, and EGR1). Furthermore, 5 miRNAs and 5 transcription factors were identified, including E2F, HSF2, and miR-26. miR-26 may participate in THCA by targeting CITED1 and PLA2R1; E2F may participate in THCA by regulating ITGA3, TIMP1, KRT19, EGR1, and JUN; HSF2 may be involved in THCA development by regulating SERPINA1 and FOSB; and small-molecule drugs may have anti-THCA effects. Our results provide novel directions for mechanistic studies and drug design of THCA.
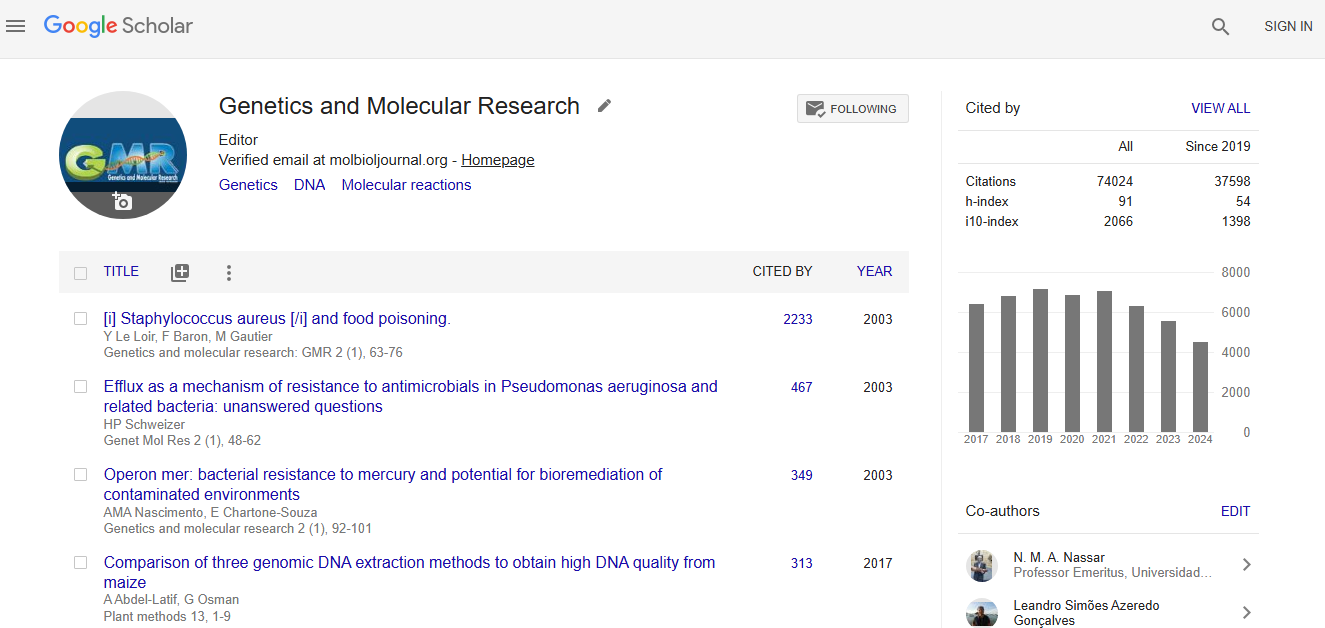
Impact Factor an Index

Google scholar citation report
Citations : 74024
Genetics and Molecular Research received 74024 citations as per google scholar report