Abstract
Comparison of gene regulatory networks of benign and malignant breast cancer samples with normal samples
Author(s): D.B. Chen H.J. YangThe aim of this study was to explain the pathogenesis and deterioration process of breast cancer. Breast cancer expression profile data GSE27567 was downloaded from the Gene Expression Omnibus (GEO) database, and breast cancer-related genes were extracted from databases, including Cancer-Resource and Online Mendelian Inheritance In Man (OMIM). Next, h17 transcription factor data were obtained from the University of California, Santa Cruz. Database for Annotation, Visualization, and Integrated Discovery (DAVID)-enrichment analysis was applied and gene-regulatory networks were constructed by double-two-way t-tests in 3 states, including normal, benign, and malignant. Furthermore, network topological properties were compared between 2 states, and breast cancer-related bub genes were ranked according to their different degrees between each of the two states. A total of 2380 breast cancer-related genes and 215 transcription factors were screened by exploring databases; the genes were mainly enriched in their functions, such as cell apoptosis and proliferation, and pathways, such as p53 signaling and apoptosis, which were related with carcinogenesis. In addition, gene-regulatory networks in the 3 conditions were constructed. By comparing their network topological properties, we found that there is a larger transition of differences between malignant and benign breast cancer. Moreover, 8 hub genes (YBX1, ZFP36, YY1, XRCC5, XRCC4, ZFHX3, ZMAT3, and XPC) were identified in the top 10 genes ranked by different degrees. Through comparative analysis of gene-regulation networks, we identified the link between related genes and the pathogenesis of breast cancer. However, further experiments are needed to confirm our results. The aim of this study was to explain the pathogenesis and deterioration process of breast cancer. Breast cancer expression profile data GSE27567 was downloaded from the Gene Expression Omnibus (GEO) database, and breast cancer-related genes were extracted from databases, including Cancer-Resource and Online Mendelian Inheritance In Man (OMIM). Next, h17 transcription factor data were obtained from the University of California, Santa Cruz. Database for Annotation, Visualization, and Integrated Discovery (DAVID)-enrichment analysis was applied and gene-regulatory networks were constructed by double-two-way t-tests in 3 states, including normal, benign, and malignant. Furthermore, network topological properties were compared between 2 states, and breast cancer-related bub genes were ranked according to their different degrees between each of the two states. A total of 2380 breast cancer-related genes and 215 transcription factors were screened by exploring databases; the genes were mainly enriched in their functions, such as cell apoptosis and proliferation, and pathways, such as p53 signaling and apoptosis, which were related with carcinogenesis. In addition, gene-regulatory networks in the 3 conditions were constructed. By comparing their network topological properties, we found that there is a larger transition of differences between malignant and benign breast cancer. Moreover, 8 hub genes (YBX1, ZFP36, YY1, XRCC5, XRCC4, ZFHX3, ZMAT3, and XPC) were identified in the top 10 genes ranked by different degrees. Through comparative analysis of gene-regulation networks, we identified the link between related genes and the pathogenesis of breast cancer. However, further experiments are needed to confirm our results.
Impact Factor an Index

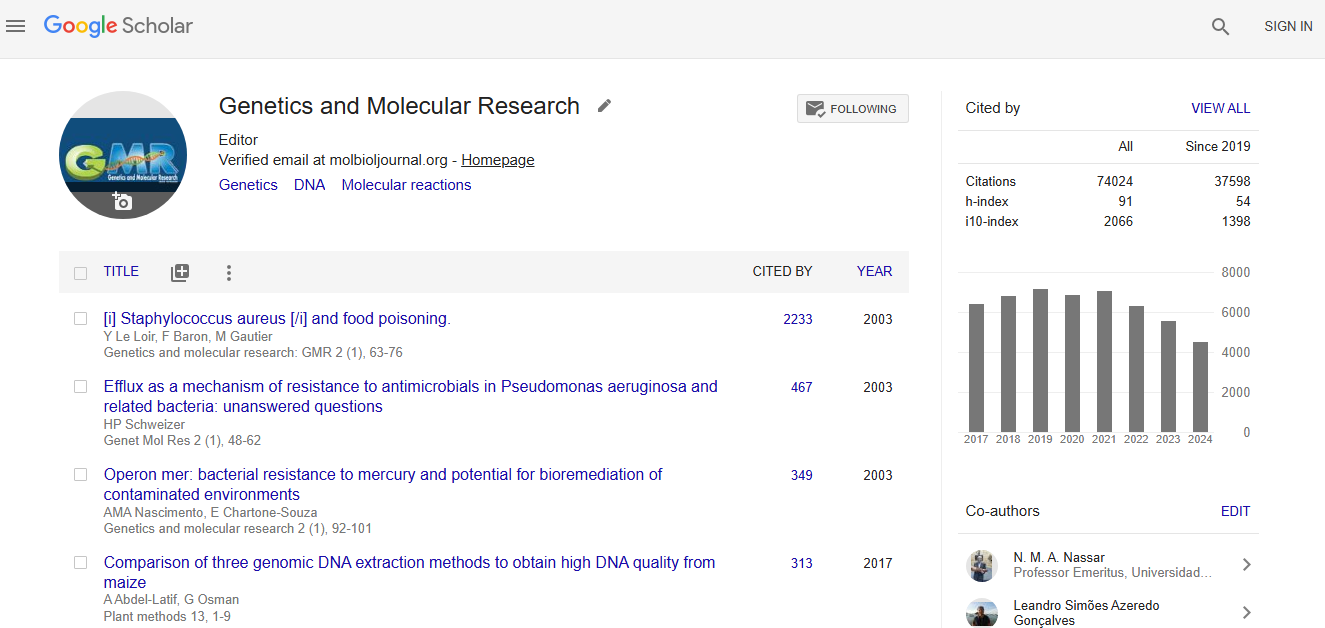
Google scholar citation report
Citations : 74024
Genetics and Molecular Research received 74024 citations as per google scholar report