Abstract
Clinical features of Mexican patients with Mucopolysaccharidosis type I
Author(s): A. Alonzo-Rojo, J.E. GarcÃ?Âa-Ortiz, M. Ortiz-Aranda, M.P. Gallegos-Arreola and L.E. Figuera-VillanuevaMucopolysaccharidosis type I (MPS-I) is an autosomal recessive lysosomal storage disorder caused by a deficiency or absence of α-l-iduronidase, which is involved in the catabolism of glycosaminoglycans (GAGs). This deficiency leads to the accumulation of GAGs in several organs. Given the wide spectrum of the disease, MPS-I has historically been classified into 3 clinical subtypes - severe (Hurler syndrome), intermediate (Hurler-Scheie syndrome), and mild (Scheie syndrome) - none of which is determined by residual enzyme activity. Eleven Mexican patients with MPS-I from northwestern México were evaluated. Diagnoses were confirmed through quantification of GAGs in urine and enzyme assay for α-l-iduronidase. Regardless of phenotype, all patients had various degrees of infiltrated facies, short stature, dysostosis multiplex, joint contractures, and corneal opacity typical of the disease. A better understanding of the spectrum of this disease can assist in diagnosis, treatment, and improvement in the quality of life for these patients.
Impact Factor an Index

Google scholar citation report
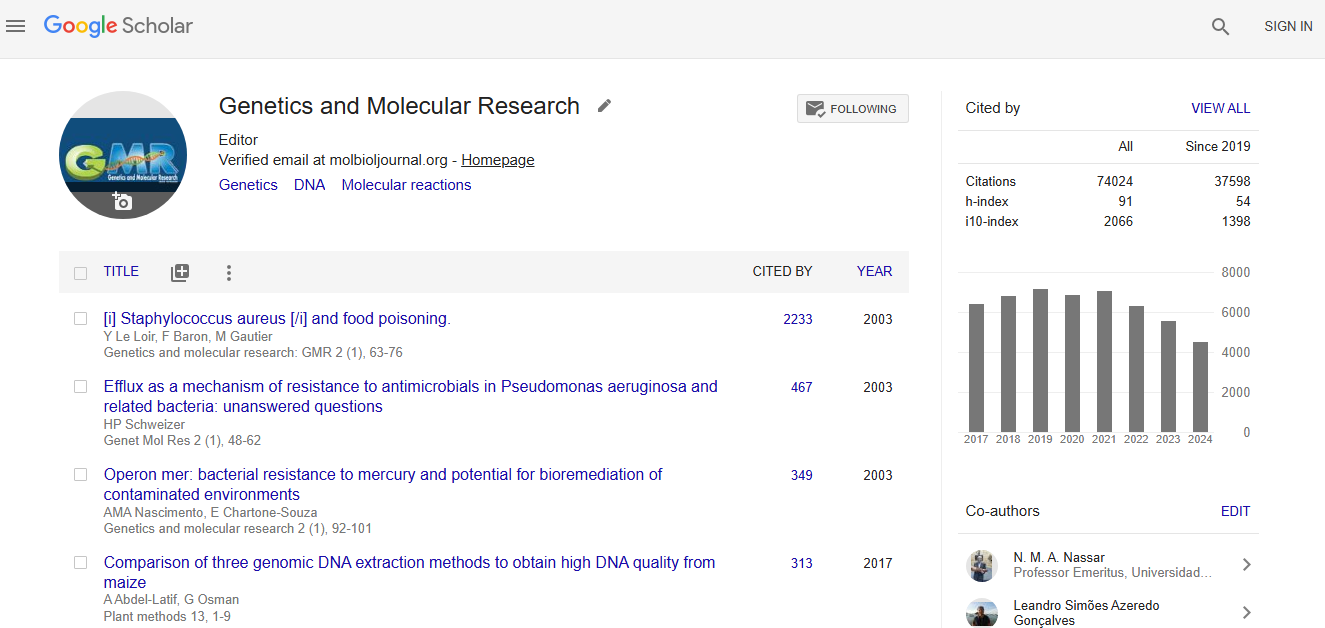
Citations : 74024
Genetics and Molecular Research received 74024 citations as per google scholar report