Abstract
Bioinformatic analyses of GRAS genes in Betula kirghisorum based on transcriptome data
Author(s): C.J. Yang, G.Y. Li and Y.L. CuiThe transcriptomes of salt-stressed and unstressed Betula kirghisorum plants were analyzed using high throughput sequencing technology. A total of 52,239,804 and 51,772,998 clean reads were obtained from the two libraries, respectively, and de novo assembled into 60,545 all-unigenes. A total of 39,997 unigenes were annotated using public databases. Overall, 7206 genes were differentially expressed in unigenes and were involved in 127 pathways. Thirteen transcription factor families were identified in B. kirghisorum, including GRAS proteins, which are plant-specific transcription factors. By using bioinformatic methods to predict and analyze physicochemical properties, structural data were obtained on the 19 potential GRAS proteins. The results revealed that these proteins are hydrophilic, with significant differences in their length and molecular weight. The main secondary structures were alpha helices and random coils. BkGRASproteins possess typical GRAS domains: LHR I; VHIID motif; LHR II; PFYRE motif; and SAW motif. In the majority of BkGRAS proteins, AGG, AGA, UCU, GCU, GGG, CCA, GUU, GUG, AUU, GAU, and AAG codons were used preferentially. Aside from the BkGRAS17 gene (relative synonymous codon usage (RSCU) = 1.20), usage of the UUA codon by other BkGRAS genes was low (RSCU < 1.0). The effective number of codons showed that BkGRAS genes have low codon bias. Subcellular localization analysis that predicted these proteins are found in the nucleus, cytoplasm, or chloroplast. BkGRAS proteins were divided into six subfamilies: SCR, LISCL, SCL3, DELLA, HAM, and PAT1. These results provide important information for the further functional study of GRAS genes in B. kirghisorum.
Impact Factor an Index

Google scholar citation report
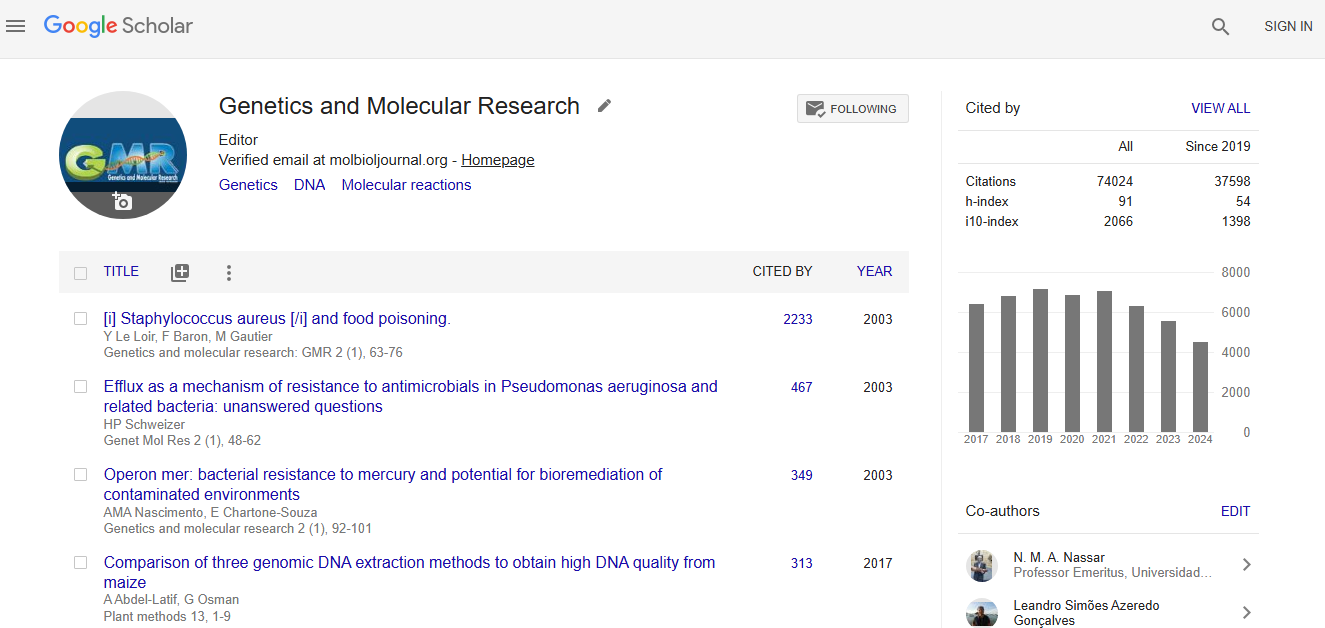
Citations : 74024
Genetics and Molecular Research received 74024 citations as per google scholar report