Abstract
A novel homozygous stop gain mutation in SLC12A3 gene cause Gitelman syndrome in Saudi consanguineous family
Author(s): Muhammad Imran Naseer, Osama Yousef Muthaffar , Mahmood Rasool, Angham Abdulrahman Abdulkareem, Mohammad Alam Jafri, Peter Natesan Pushparaj, Gauthaman Kalamegam, Adeel G. Chaudhary, Mohammad H. Al-QahtaniGitelman syndrome (GS) is a genetic disorder that affects kidney and causes an imbalance of charged atoms (ions) in the body, including ions of potassium, magnesium, and calcium. GS is characterized by hypokalemia and metabolic alkalosis. GS is a rare autosomal recessive renal tubulopathy disease caused by loss-of-function mutations in the SLC12A3 gene. Objective of the present study is to find the genetic causes involved in a consanguineous Saudi family. Whole exome sequencing was done to find out the genetic mutation in the affected members of the family and the putative mutation was further validated by subsequent Sanger sequencing in other member of the family. We identified a novel homozygous mutation c.2686C>T as a result in p.Arg896* stop gained in exon 23 of SLC12A3 gene. The mutation was ruled out in 100 unrelated healthy controls. A novel homozygous stop gain mutation detected in this study has not yet been reported as pathogenic in literature or variant databases. Functional prediction of this mutation was performed using various online in silico prediction tools and all predicted it be disease causing. In conclusion, the here detected homozygous SLC12A3 mutation in the SLC12A3 gene as a result a truncated protein produced with a premature stop gain would expected to lead to functional loss to cause GS in Saudi family. This study will add more knowledge about GS in Arab populations’ database of genetic disorders and will help in the genetic counselling of the families in Saudi Arabia.
Impact Factor an Index

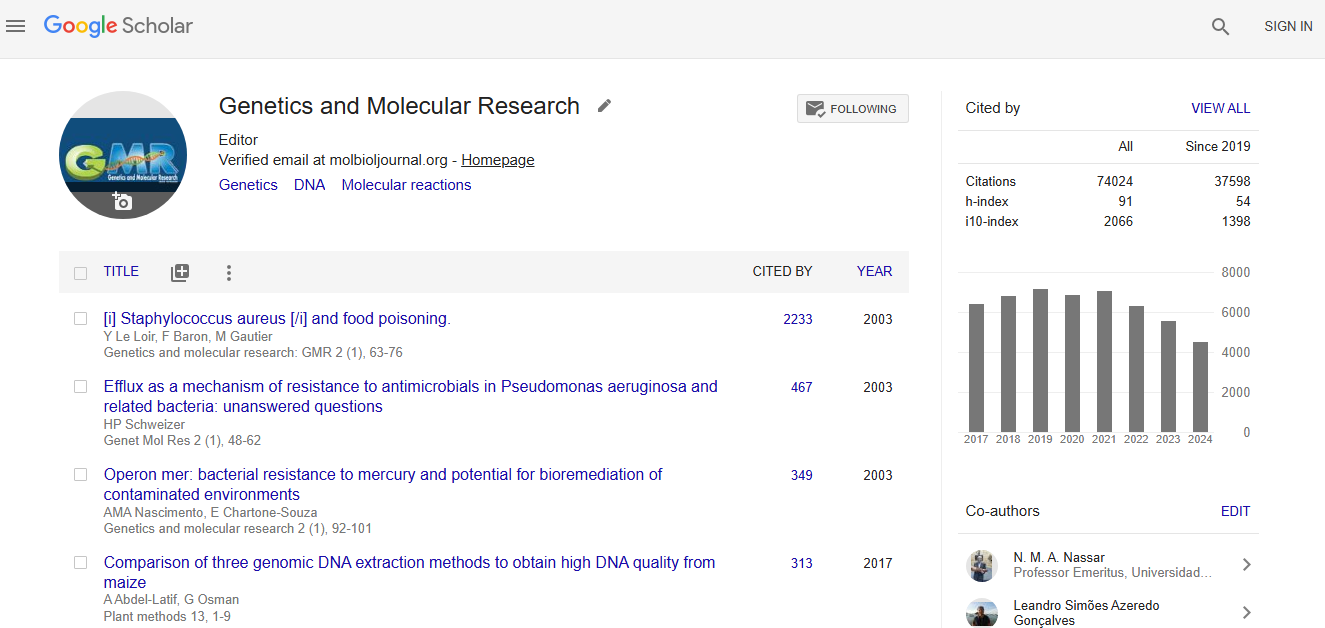
Google scholar citation report
Citations : 74024
Genetics and Molecular Research received 74024 citations as per google scholar report